

Post-transcriptional processing of precursor mRNAs (pre-mRNAs) greatly contribute to enhance transcriptome diversity, coding capacity of a genome and multiple gene regulatory mechanisms in eukaryotes. Characterization of transcriptome diversity has been extensively dominated by Second-generation sequencing technologies (NGS), driven by its superiority in producing highly accurate (>99.9%) sequencing reads, on a massive scale with cost-effectiveness. Yet, the optimal application of it to precisely predict full-length transcript isoforms is challenging with its short-read length. In addition, many reference transcript annotations are still based on short-read RNA-seq, which are thereby incomplete.

While with the advent of PacBio Single-Molecule Real-Time (SMRT) technology, developed by Pacific BioSciences (PacBio), high-quality long reads across the entire transcriptome offers solutions to these shortcomings by sequencing full-length mRNA transcripts from 5’UTR to 3’poly-A tail, without assembly.
Here, we present a paper titled ‘‘A survey of the sorghum transcriptome using single-molecule long reads’’ conducted by Salah E. Abdel-Ghany, Michael Hamilton, Jennifer L. Jacobi, et al. from Program in Cell and Molecular Biology, Colorado State University. Here, the researchers integrated PacBio Isoform Sequencing (Iso-Seq) to transcriptome analysis on sorghum transcriptome, to identify full-length splice isoforms and alternative polyadenylation (APA) sites. This Paper provided the first comprehensive view of splice variants in sorghum, underscoring the advantage of Iso-Seq in identifying full-length splice isoforms.

Despite the fact that several large-scale RNA-seq studies have been performed in plants to analyse AS, currently it is not known how many distinct splice isoforms are produced.


The researchers developed a pipeline called TAPIS (Transcriptome Analysis Pipeline for Isoform Sequencing) to identify full-length splice isoforms and APA sites and serve as a useful tool to analyze Iso-Seq data from any organism.
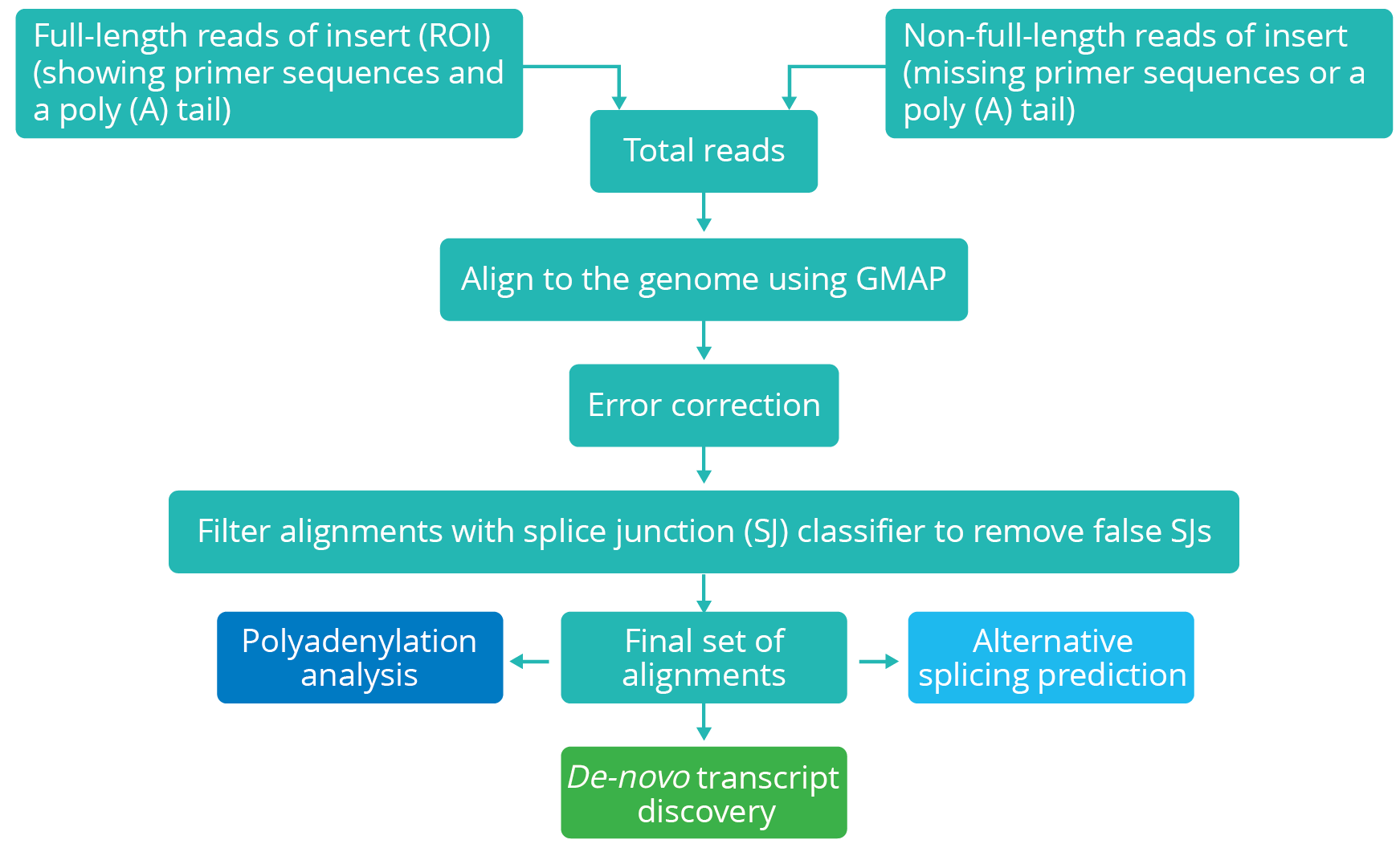


Identification of transcriptome-wide full-length isoforms in sorghum at an unprecedented scale with over 11,000 novel splice isoforms
Detection of B11,000 new splicing events from the Iso-Seq reads as compared with splicing events in the gene models.
Uncovering of extensive APA of sorghum transcripts (B50% of expressed genes were found to have multiple polyadenylation sites)
Identification of over 2,100 novel genes that were not previously annotated. Among the novel genes, many are putative long non-coding transcripts.

The research greatly enhance sorghum gene annotations and aid in studying gene regulation in this important bioenergy crop. The TAPIS pipeline will serve as a useful tool to analyse Iso-Seq data from any organism.


[1] Chang Y, Hu T, Zhang W, Zhou L, Wang Y, Jiang Z. Transcriptome profiling for floral development in reblooming cultivar ‘High Noon’ of Paeonia suffruticosa. Sci Data. 2019 Oct 22;6(1):217. doi: 10.1038/s41597-019-0240-1. PMID: 31641161; PMCID: PMC6805890.


Novogene Corporation Inc.
![]() 916-252-0068-383
916-252-0068-383
![]() inquiry_us@novogene.com
inquiry_us@novogene.com
![]() www.novogene.com
www.novogene.com
Copyright©2011-2023 Novogene Corporation
All Rights Reserved. Information and specifications are subject to change at any time without notice.